Muscular dystrophies are genetic diseases that cause progressive muscle weakness. The best known is that described by Duchenne, which affects boys, and which results from mutations in the gene encoding a protein called dystrophin. Another subclass is the congenital muscular dystrophies, where muscle weakness is apparent at birth or shortly afterwards. One of them is muscle-eye-brain disease (MEB). Children carrying the faulty MEB gene suffer from both muscle weakness and cobblestone lissencephaly, in which a flaw in neuronal migration results in a brain with a bumpy, cobblestone appearance and loss of the normal folding pattern. Recently, we identify a gene of glycosyltransferase is responsible for MEB and we propose that interference in glycosylation is a new pathomechanism for muscular dystrophy as well as neuronal migration disorder.
α-Dystroglycan is an extracellular peripheral membrane glycoprotein anchored to the cell membrane by binding to a transmembrane glycoprotein, β-dystroglycan. These two dystroglycan subunits were originally identified as members of the sarcolemmal dystrophin-glycoprotein-complex (DGC). α- and β- dystroglycans are encoded by a single gene, and cleaved into separate proteins by posttranslational processing. The α-dystroglycan-β-dystroglycan complex is widely expressed in a broad array of tissues and thought to act as a transmembrane linker between the extracellular matrix and intracellular cytoskeleton, because α-dystroglycan binds to laminin, and the intracellular domain of β-dystroglycan interacts with dystrophin in skeletal muscle.
α-Dystroglycan is heavily glycosylated, and we demonstrated that the sialic acid residues, which are probably attached to O-linked oligosaccharides, were essential for laminin-binding. Further, we have elucidated the structure of O-linked oligosaccharides of bovine peripheral nerve α-dystroglycan as a novel O-mannosyl glycan, Siaα2-3Galβ1-4GlcNAcβ1-2Man. In another study, we found the same O-mannosyl glycan in rabbit skeletal muscle α-dystroglycan. After our report on the O-mannosyl glycan, a series of mammalian type O-mannosyl glycans have been found. In summary, O-mannosylation is known as a yeast-type modification, and oligomannose-type O-mannosylated glycoproteins are abundant in the yeast cell wall. On the other hand, mammalian O-mannosylation is a rare type of protein modification that is observed in a limited number of glycoproteins of brain, nerve, and skeletal muscle. Future studies are needed to clarify the distribution of such O-mannosyl glycans in various tissues.
Analysis of the biosynthetic pathway of the O-mannosyl glycans in mammals is important for elucidating not only the regulation of expression but also the biological functions of these glycans. The identification and characterization of the enzymes involved in the biosynthesis of mammalian type O-mannosyl glycans will be an important step forward elucidating these glycans. A key difference between mammalian and yeast-type O-mannosyl glycans is that those in mammals have the GlcNAcβ1-2Man linkage. This linkage is assumed to be catalyzed by a glycosyltransferase, UDP-N-acetylglucosamine: protein O-mannose β1,2-N-acetylglucosaminyltransferase (POMGnT1). POMGnT1 catalyzes the transfer of N-acetylglucosamine from UDP-GlcNAc to O-mannosyl glycoproteins, according to the reaction: UDP-GlcNAc + Man-R ¨ GlcNAcβ1-2Man-R + UDP in which R is protein. After we succeeded in developing an enzyme assay for POMGnT1, its activity was found in brain homogenates of several mammals. It should be noted that GlcNAcβ1-2Man linkages are also found in N-glycans, where they are catalyzed by two enzymes, UDP-N-acetylglucosamine: α-3-D-mannoside β-1,2-N-acetylglucosaminyltransferase I (GnT-I) and UDP-N-acetylglucosamine: α-6-D-mannoside β-1,2-N-acetylglucosaminyltransferase II (GnT-II). However, we found that recombinant GnT-I and GnT-II had no ability to catalyze the GlcNAcβ1-2Man linkage in O-mannosyl glycans, suggesting that a new enzyme must be responsible for the formation of this linkage. Thus, we cloned the human POMGnT1 gene on the basis of human cDNA sequences homologous to human GnT-I.
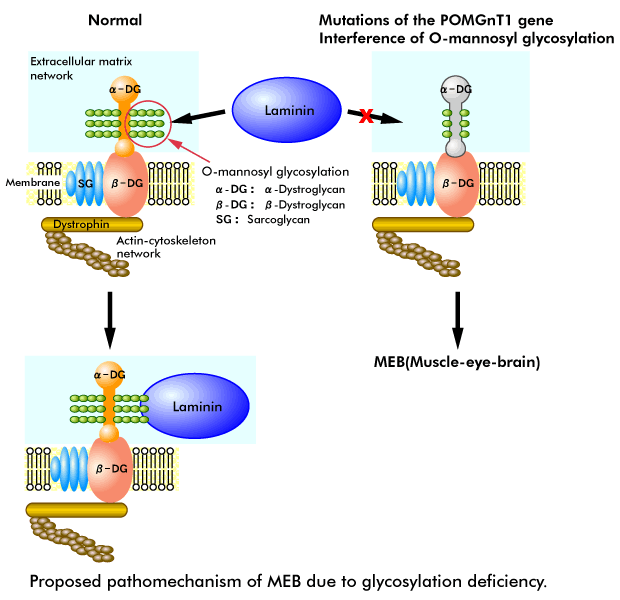
The human POMGnT1 gene exists at 1p33, and we find that the POMGnT1 gene is located within the small candidate interval for muscle-eye-brain disease (MEB: OMIM 253280). MEB is an autosomal recessive disorder characterized by congenital muscular dystrophy, ocular abnormalities and brain malformation (type II lissencephaly). Patients with MEB show congenital muscular dystrophy, severe congenital myopia, congenital glaucoma, pallor of the optic discs, retinal hypoplasia, mental retardation, hydrocephalus, abnormal electroencephalograms and myoclonic jerks. All infants with MEB are floppy with generalized muscle weakness, including facial and neck muscles, from birth. Muscle biopsies show dystrophic changes, and brain MRIs reveal pachygyria-type cortical neuronal migration disorder, flat brainstem and cerebellar hypoplasia. Since defects of DGC cause muscular dystrophies and O-mannosyl type glycan is required for the laminin binding of α-dystroglycan in DGC, it is possible that mutations in the POMGnT1 gene are related to MEB.
To test this hypothesis, we screened the whole coding region and the exon/intron flanking sequences of the POMGnT1 gene for mutations in six patients with MEB. We identified six independent disease-causing mutations in these patients and we have not detected these six substitutions in any of 246 normal chromosomes, indicating that these mutations are pathogenic and that the POMGnT1 gene is responsible for MEB. To confirm that the mutations observed in patients with MEB are responsible for the defects in the synthesis of O-mannosyl glycan, we expressed the most frequent mutation and found a loss of enzymatic activity. Additionally, we found a selective deficiency of α-dystroglycan in MEB patients. This finding suggests that α-dystroglycan is a potential target of POMGnT1 and that altered glycosylation of α-dystroglycan may play a critical role in the pathomechanism of MEB.
Recent investigations have revealed that some muscular dystrophies may be caused by abnormal glycosylation of α-dystroglycan including Fukuyama-type congenital muscular dystrophy, MDC1C, and the myodystrophy (myd) mouse, although the detailed defect is still unclear. Identification of these defects provides new directions for unraveling a glycopathomechanism for muscular dystrophy.